Introduction
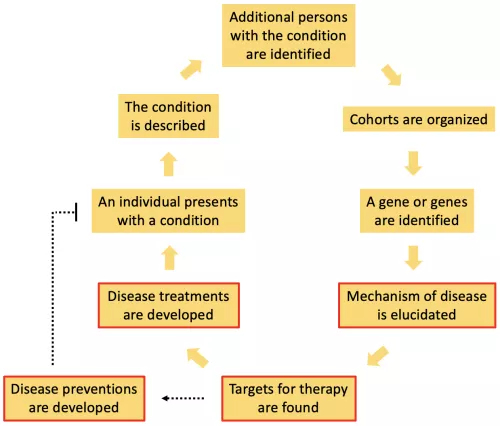
Common diseases that generally are not passed on by Mendelian genetics can be better understood and treated by taking advantage of the fact that there are rare familial forms of the disease. For example, most cases of Alzheimer's Disease are sporadic, or not caused by a single genetic mutation. However, there are families in which members develop the disease at much higher rates than the general population, and from these families or cohorts, we have learned about genetic variants in APP, presenilin and ApoE that predispose to disease. In our lab, we are interested in all parts of the process of describing, identifying and researching diseases, but we are most actively engaged in the parts of the process that are highlighted in red. Specifically, we study how variations in the progranulin, MAPT/tau and newly identified genes perturb cellular functions, leading to particular disease phenotypes. To do so, we have taken advantage of the strong molecular genetic techniques available in C. elegans and then confirmed and extended our findings in mammalian models. Our goal is to translate our findings into treatments and even preventions for neurodegenerative diseases like Alzheimer's Disease, frontotemporal dementia and related disorders.
Progranulin, granulins and regulation of cellular stress responses

Mutations in progranulin lead to the development of frontotemporal lobar degeneration, a devastating neurodegenerative disease. Disease-related progranulin mutations result in a genetic haploinsufficiency, or production of progranulin protein levels that are approximately half that of normal. Apoptosis or programmed cell death is a normal process of removal of excess, unnecessary or damaged cells. Using the nematode C. elegans and cultured murine cells, we discovered that loss of progranulin affects the rate of clearance of apoptotic cells. Why is this? One project in the lab is to better understand how progranulin regulates programmed cell death kinetics and how progranulin deficiency leads to neurodegeneration. Additional projects involve the function of granulins, the cleavage fragments of progranulin, in regulating protein homeostasis and lysosome function.
Pathophysiological implications of the MAPT mutations

Mutations in the MAPT or tau gene have been associated with several neurodegenerative diseases and tau "tangles" are found in the brains of those afflicted with Alzheimer's Disease. Using patient-derived fibroblasts, induced pluripotent stem cells (iPSCs) and other cellular models, we are studying the effects of these mutations and newly described polymorphisms, on tau function. We are also utilizing CRISPR/Cas gene editing techniques in human induced pluripotent stem cells to explore the influence of disease-associated polymorphisms on genotype-phenotype interactions.
Dysregulation of pH dynamics in neurodegenerative disease and aging

Recent failures in Alzheimer Disease (AD) drug trials have highlighted the need for better understanding of the basic cell and molecular biology underlying AD and other neurodegenerative disorders. Although it is clear that intracellular pH becomes more acidic in AD, until now this has been considered a consequence of programmed cell death activation rather than a driving force in disease. However, pH changes can precede and occur independently of cell death programs. We have seen that granulin levels can impact cellular pH. Thus, we hypothesize that dysregulation of cellular pH actively promotes neuronal dysfunction and death in Alzheimer’s Disease by preventing protein clearance and promoting protein aggregation. In this highly collaborative project, we will interrogate pH in disease models by both measuring and manipulating cytosolic and lysosomal pH. The insights gained from these studies have the potential to fundamentally alter our understanding of the disease process and could lead to key adjunctive therapies in a broader strategy to treat and prevent AD.
Elucidating regulatory nodes in tau metabolism and pathobiology

Genetic mutations in tau and other pathway members can disrupt tau metabolism, leading to tau accumulation, secretion, and neurodegeneration. We will generate critically important information about tau homeostasis and a foundational basis from which to build and frame subsequent investigations into tau pathobiology and toxicity. Our hypothesis is that proper tau metabolism requires the precise, coordinated action of molecular chaperones, co-chaperones, post-translational modifications (PTMs), and degradation machinery that each represent regulatory nodes. To learn more about our research with our FTD CWOW team, please visit the CWOW homepage.
Fein Memory and Aging Center Fibroblast Bank

The Fein Memory and Aging Center (MAC) Fibroblast Bank provides access to patient-derived fibroblast cell lines to support research in neurodegenerative disease and related fields. Our repository includes curated fibroblast samples available to qualified investigators for research purposes.
If you are interested in requesting fibroblast cell lines or would like additional information about available samples, please contact Dr. Aimee Kao.